 Атаксия Фридрейха — наследственное нейродегенеративное заболевание, для которого характерно нарушение выведения ионов железа из околомитохондриального пространства клетки.
Атаксия Фридрейха — наследственное нейродегенеративное заболевание, для которого характерно нарушение выведения ионов железа из околомитохондриального пространства клетки.
Среди европейцев распространенность заболевания составляет 1:20 000–1:50 000, а во всем мире каждый 120-й житель имеет предрасположенность к данной патологии. Причиной развития атаксии Фридрейха является мутация в гене FXN, в частности, нестабильное увеличение триплетов GAA. Данный ген кодирует специфический белок фратаксин, который отвечает за транспорт ионов железа из околомитохондриального пространства и не допускает тем самым образования свободных радикалов, которые оказывают выраженное повреждающее действие на центральную и периферическую нервную систему, а также другие органы.
Наследование атаксии Фридрейха проходит по аутосомно-рецессивному типу. Возможно бессимптомное носительство гена.
Причины развития атаксии Фридрейха
Развитие заболевания связано с дисбалансом внутриклеточного железа, его высокая концентрация в митохондриях вызывает увеличение свободных радикалов, разрушающих клетку. Дисбаланс возникает при недостаточности или искажении структуры белка, синтезируемого в цитоплазме – фратаксина. Этот белок отвечает за транспорт железа из митохондрий, при накоплении которого свыше нормы происходит снижения цитозного железа.
Это главные причины развития атаксии Фридрейха, в результате которых активируются гены, кодирующие ферроксидазу и пермеазу, которые, как и фратаксин отвечают за транспорт железа. Это приводит к еще большему накоплению его в митохондриях. Наследственность обусловлена так называемым геном болезни Фридрейха предположительно обнаруживающийся в центномерной области 9-й хромосомы в локусе 9ql3 — q21. Могут происходить несколько мутаций одного гена, чем, и обусловлены разные формы заболевания. Атаксия Фридрейха занимает половину случаев возникновения атаксий. Первые признаки проявляются в возрасте до 20 лет, значительно реже до 30. Возникает одинаково часто и у женщин и у мужчин, не подвергаются этой болезни только представители негроидной расы
Заболевание поражает нейроны центральной и периферической нервной системы, но медицина не имеет объяснения, причины, почему в нервной системе повреждаются только проводящие пути спинного мозга. В других системах заболеванию подвергаются не менее важные клетки органов, это клетки миокарда, β — клетки островков Лангерганца в поджелудочной железе, клетки сетчатки и костных тканей.
Течение заболевания постоянно прогрессирующее. Если отсутствует адекватное лечение атаксии Фридрейха, длительность болезни не превышает 20 лет. И начав проявляться неловкостью и неуверенностью при ходьбе, дизартрией, через некоторое время полностью лишает человека нормально координировать движения и передвигаться самостоятельно. Заболевание оканчивается летально, в редких случаях при отсутствии таких проявлений как сахарный диабет и болезни сердца больные доживают до 70-80 лет.
1.Общие сведения
Атаксия (древнегреч. «неупорядоченность») – неврологический патологический феномен, заключающийся в рассогласованности, хаотичности моторики: нарушаются сложные нейромышечные взаимосвязи, в результате чего движения больного становятся нецеленаправленными и непродуктивными. Атаксия, выраженная в той или иной степени, сопутствует многим заболеваниям центральной нервной системы (ЦНС), – например, атрофическим и опухолевым, а также некоторым психопатологическим расстройствам, – однако выделяется группа атаксий как самостоятельных болезней.
В частности, известно более двух десятков вариантов наследственной спиноцеребеллярной (т.е. обусловленной сочетанным поражением спинного мозга и мозжечка) атаксии, или SCA, представляющей собой генетически обусловленный дегенеративно-дистрофический процесс в указанных структурах ЦНС.
Семейная атаксия Фридрейха (названа по имени врача, который в 1860 году дал первое клиническое описание болезни) является наиболее распространенным из наследственных нейродегенеративных заболеваний с преобладанием атаксии в клинической картине. Встречается, по различным источникам и в некоторой региональной зависимости, у 3-7 человек на каждые сто тысяч населения; пассивное носительство дефектного гена распространено гораздо шире (около 1%). От пола вероятность запуска процесса не зависит.

Обязательно для ознакомления! Помощь в лечении и госпитализации!
Симптомы атаксии Фридрейха
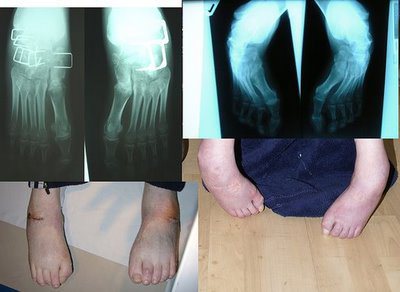
Первые симптомы заболевания заключаются в угнетении ахилловых и коленных рефлексов. Эти симптомы появляются за несколько лет до появления других, так же к раним проявлениям, относится ревмокардит, который часто лечится как отдельное заболевание. Так что не считается, что это симптомы атаксии Фридрейха до появления неврологических нарушений. Происходят постепенные скелетные деформации, такие как сколиоз, деформация пальцем рук и ног, «стопа Фридрейха», при которой наблюдается ненормальное разгибание пальцев в основных фалангах, а стопа имеет высокий вогнутый свод.
Атаксия Фридрейха в развернутой форме характеризуется типичными для атаксий неврологическими нарушениями и тотальной арефлексией. Нарушаются суставно-мышечная и вибрационная чувствительности, мышечная гипотония, симптом Бабинского. Постепенно развивается сенситивная и мозжечковая атаксии, атрофия и слабость мышц ног.
В 90% больных проявляются экстраневральные проявления, это поражения сердца, эндокринные расстройства, катаракта. Развивается прогрессирующая кардиомиопатии, она может быть как гипертрофической, так и дилатационной. В этом случае наблюдаются такие симптомы атаксии Фридрейха, как боли в области сердца, сердцебиение, систолические шумы, одышка. Характерны эндокринные заболевания, такие как сахарный диабет, гипогонадизм, инфантилизм, дисфункция яичников.
Поздняя стадия атаксии характеризуется амитрофией и расстройством глубокой чувствительности, исчезновением сухожильных и надкостных рефлексов. Что распространяется на верхние конечности. Происходит глубокий распад моторных функций, из-за чего человек теряет способность ходить и обслуживать себя. Развивается кифосколиоз с формированием горба, деформация кистей рук. Из экстраневральных проявлений может, наблюдается нистагм, снижение слуха, атрофия зрительных нервов, нарушение функции тазовых органом, деменция. Прогрессирующая кардиомиопатия на поздних стадиях заболевания является причиной смерти у половины больных, чаще всего из-за нарушений в проводной системе сердца. К непосредственным причинам смерти так же относятся легочная недостаточность и инфекционные осложнения.
Прогноз заболевания
Учитывая отсутствие патогенетического лечения, следует понимать, что заболевание носит неуклонно прогрессирующий характер. Пациенты с диагностированной болезнью в среднем живут 10-15 лет и как правило не более 30 лет с момента начала ее развития, у женщин течение заболевания носит более благоприятный характер – длительность их жизни нередко превышает 20 лет. Летальный исход наступает в результате развития легочной или сердечной недостаточности.
Однако своевременная диагностика и применение вышеуказанных методов лечения, воздействие на сопутствую патологию, недопущение развития осложнений способствуют улучшению качества жизни и ее продлению у пациентов с атаксией Фридрейха.
Диагностика атаксии Фридрейха
Компьютерная томография головного мозга, которая остается основной диагностикой атаксий при этом заболевании малоэффективная, ряд изменений можно обнаружить только на поздних стадиях. Это связано со спинальной локализацией изменений, поэтому удается обнаружить только слабую степень атрофии мозжечка на ранней стадии и атрофию полушарий, расширение стволовых цистерн, боковых желудочков и субарахноидального пространства обоих полушарий на более поздних стадиях. Ранняя диагностика атаксии Фридрейха производится с помощью МР-томографии, которая дает возможность обнаружить атрофию спинного мозга, а на развернутой стадии, и умерено выраженную атрофию моста, мозжечка и продолговатого мозга. На начальной стадии обязательно проводится электрофизиологическое исследование, при таких исследованиях устанавливается тяжесть поражения чувствительности нервов конечностей.
Для полной диагностики проводят нагрузочные тесты толерантности к глюкозе, рентгеновское исследование позвоночника. В первую очередь диагностика направлена на то что бы точно, установить диагноз и дифференцировать заболевание от других со схожей симптоматикой. К примеру симптомы атаксии Фридрейха могут быть такими же как и наследственной атаксии при дефиците витамина Е, синдрома Бассена-Корнцвейга, наследственных обменных заболеваний, таких как болезнь Краббе и болезнь Ниманна-Пика. Схожая симптоматика может быть и при рассеянном склерозе, за исключением сухожильной арефлексии, мышечной гипотонии и экстраневральных проявлений. Не характерно для атаксии Фридрейха наличие ремиссий и изменения плотности вещества мозга, что наблюдается при диагностике рассеянного склероза.
- Читайте также:
Для дифференцирования заболевания назначается и ряд дополнительных лабораторных исследований. Проводится ДНК тестирование и медико-генетическое консультирование, исследование липидного профиля крови, анализ мазка крови на наличие дефицита витамина Е и акантоцитов. Лечение атаксии Фридрейха не приводит к полному выздоровлению, но своевременная профилактика дает возможность избежать развития многих симптомов и осложнений. Диагностика атаксии Фридрейха с помощью ДНК тестирования должна назначаться не только больному, а и родственникам для определения наследственности заболевания, это необходимо в целях профилактики, и назначения превентивной терапии.
4.Лечение
Этиопатогенетической терапии или средств профилактики на сегодняшний день нет; все принимаемые меры (даже радикальные, например, ортопедическое хирургическое вмешательство) носят лишь паллиативный характер и направлены на смягчение доминирующей симптоматики. Прогноз, в целом, неблагоприятный: неуклонное прогрессирование нейродегенеративного процесса результирует летальным исходом через 15-20 лет от манифестных проявлений. Однако в отдельных случаях, когда пациент находится под постоянным терапевтическим контролем и удается затормозить развитие тяжелой сердечной, легочной, эндокринной недостаточности, больные доживают до старости.
Диета, питание при атаксии Фридрейха
Диета для нервной системы
- Эффективность: лечебный эффект через 2 месяца
- Сроки: постоянно
- Стоимость продуктов: 1700-1800 рублей в неделю
Диета 9-й стол
- Эффективность: лечебный эффект через 14 дней
- Сроки: постоянно
- Стоимость продуктов: 1400 — 1500 рублей в неделю
Низкоуглеводная диета
- Эффективность: От 3 до 7 кг за 2 недели
- Сроки: 14 дней
- Стоимость продуктов: 1250-1320 рублей в неделю
Диета при сахарном диабете
- Эффективность: лечебный эффект через 14 дней
- Сроки: постоянно
- Стоимость продуктов: 1300-1400 рублей в неделю
Доклад на СНК кафедры неврологии педиатрического факультета РГМУ 5.10.2006 года.
Болезнь Фридрейха исторически является первой нозологически самостоятельной формой наследственных атаксий, выделенной более 100 лет назад из общей группы локомоторной атаксии. Это сделал N. Friedreich. Очень компетентный врач и патолог, N.Friedreich интересовался всеми областями медицины, особенно неврологией. Friedreich обладал огромным умственным потенциалом, был одним из основоположников современной клинической неврологии.
В 1861 г. на съезде Общества немецких естествоиспытателей и врачей профессором медицины в Гейдельберге Николаусом Фридрейхом (N.Friedreich) впервые было сделано короткое предварительное сообщение о «хронической дегенеративной атрофии задних столбов спинного мозга».
В 1863-1877 гг. в серии своих классических работ, он описал подробную клиническую картину особой формы атаксии, которая рассматривалась им «…клинически – как хронически-прогрессивное расстройство комбинации движений, распространяющееся снизу вверх и захватывающее, в конце концов, речь при сохранении … органов чувств и мозговых функций, анатомически – как хроническая дегенеративная атрофия задних столбов спинного мозга».
В 1976 г. G.Geoffroy и соавт., а затем A.Harding (1981) внедрили в практику диагностические критерии «достоверной» атаксии Фридрейха. Эти критерии сыграли большую роль в выделении однородных клинических групп — лиц с достоверным диагнозом атаксии Фридрейха — с целью идентификации биохимических нарушений, лежащих в основе болезни.
В 1982 г. A. Barbeau и представители его школы впервые высказали предположения о роли митохондриальных нарушений в патогенезе атаксии Фридрейха и предложили вместо общепринятого тогда названия «атаксия Фридрейха» более точный термин «болезнь Фридрейха», отражающий множественность неврологических и экстраневральных проявлений заболевания.
Болезнь Фридрейха — наследственное заболевание, характеризующееся медленно прогрессирующей атаксией вследствие склеротического перерождения задних и боковых столбов спинного мозга, гипоплазией мозжечка и спинного мозга.
 Читайте также:
Читайте также:

Болезнь Фридрейха — самая частая форма наследственных атаксий, распространенность составляет 2—7 на 100000 населения. Интересно, что данное заболевание встречается практически исключительно у лиц белой расы и не наблюдается у коренных представителей негроидной расы или азиатских народов.
Генетические данные.
Болезнь Фридрейха наследуется по аутосомно-рецессивному типу.
Ген болезни Фридрейха был картирован в центромерной области 9-й хромосомы в локусе 9ql3 — q21 (Chamberlain S. et al., 1988; Hanauer A. et al., 1990). В нашей стране первый опыт косвенной ДНК-диагностики был получен сотрудниками нейрогенетического отделения НИИ неврологии РАМН совместно с лабораторией ДНК-диагностики Медико-генетического научного центра РАМН (Пугачев В.В. и др., 1996) в 1996 г.
В 1996 г. большой международной исследовательской группой был идентифицирован новый ген FRDA (другое название Х25), мутации в котором приводят к возникновению болезни Фридрейха (Campuzano V. et al., 1996). Данный ген кодирует новый митохондриальный белок фратаксин, расположенный на внутренней поверхности мембраны митохондрий и участвующий в энергетическом метаболизме клетки (в частности, регулирующий внутриклеточный обмен железа). Сущность мутации при болезни Фридрейха заключается в экспансии тринуклеотидных повторов (т.е. патологическом увеличении числа копий).
В норме в одном из участков гена содержится от 7 до 22 тандемных повторов гуанин-аденин-аденин (ГАА), а при БФ обе копии гена характеризуются увеличением (экспансией) ГАА-повторов с числом копий триплетов от 120 до 1700. Мутации такого типа носят название «динамических» в силу того, что патологически удлиненный аллель является генетически нестабильным и способен к дальнейшей экспансии при его передаче в следующее поколение.
N.Sakamoto и соавт. (1999) установили, что мутировавший участок ДНК образует уникальную, отличную от нормальной бимолекулярную структуру — так называемую «липкую» ДНК («sticky DNA»), для образования которой требуется не менее 59 копий следующих друг за другом GAA-повторов. Таким образом, образование «липкой» ДНК может быть тем механизмом, который объясняет угнетение экспрессии гена и редукцию уровня фратаксина. В настоящее время рассматриваются три основные и взаимосвязанные между собой функции фратаксина:
1) участие белка в депонировании и выводе железа из митохондрий;
2) участие в синтезе Fe-S-белков;
- Читайте также:
3) защитная функция в отношении окислительного повреждения нейронов (Cavadini P. et al., 2002).
Результат мутации:
снижение уровня нормального фратаксина в тканях;
аккумуляция железа внутри митохондрий (Pandolfo M., 2001)
необратимое повреждение целостности и функции митохондрий;
нарушение процессов окислительного фосфорилирования;
гибель клеток энергозависимых мишеней (ЦНС –» скелетные мышцы, сердце –» орган зрения –» органы эндокринной системы –» почки –» печень).
Биохимические процессы в системе окислительного фосфорилирования и дыхательной цепи митохондрий являются мощным источником свободнорадикальных форм кислорода в клетке. При этом металлы переменной валентности, в том числе железо, играют важную роль в катализе реакций с образованием активных форм кислорода. Резкое усиление окислительных процессов при недостаточности или истощении системы антиоксидантной защиты приводит к развитию окислительного стресса, который в настоящее время рассматривается как один из общих механизмов повреждения тканей организма.
Таким образом, аккумуляция железа внутри митохондрий при болезни Фридрейха увеличивает продукцию свободных радикалов и митохондрии утрачивают способность эффективно осуществлять окислительное фосфорилирование. Следствием этого является снижение синтеза АТФ, а также дисфункция и необратимое повреждение клетки в условиях системного энергетического дефицита. Известно, что Fe-S-белки (в том числе аконитаза) чрезвычайно чувствительны к повреждающему действию свободных радикалов, поэтому количество их в клетке уменьшается. У больных формируется своеобразный порочный круг: окислительный стресс непосредственно вызывает недостаточность Fe-S-белков, а высвобождающееся при этом железо, в свою очередь, усиливает уже существующую перегрузку железом.
Таким образом, имеются два основных независимых друг от друга механизма клеточной токсичности при болезни Фридрейха — окислительный стресс и дефицит Fe-S-содержащих белков.
С этих позиций генетически, болезнь Фридрейха рассматривается как особая разновидность митохондриальной болезни, обусловленная повреждением ядерного гена. Патоморфологически же относится к спинальным формам наследственных атаксий.
Степень экспансии повторов в мутантном гене Болезни Фридрейха коррелирует с возрастом начала заболевания, особенностями его течения и клинической картины.
Патоморфология.
Прогрессирование патологического процесса при БФ, связано с гибелью нейронов в некоторых системах от периферии к центру, с возможным исчезновением тела клетки. Болезнь Фридрейха характеризуется дегенерацией задних и боковых столбов спинного мозга, особенно в люмбосакральных сегментах. Пучки Голля поражаются в большей степени, чем пучки Бурдаха. Происходит гибель клеток столбов Кларка и начинающихся от них дорсальных спиноцеребеллярных трактов, а также (обычно в поздней стадии болезни) дегенерацией ядер III, V, IX —X, XII пар черепных нервов, клеток Пуркинье, зубчатого ядра и верхней ножки мозжечка, изменения могут обнаруживаться и в больших полушариях мозга. Во всех этих областях выявляются аксональная дегенерация, демиелинизация и компенсаторный глиоз. Клеточную гибель и глиоз также можно заметить в вестибулярной и слуховой системах. При гистохимическом исследовании задних столбов спинного мозга определяется уменьшение гликолитических ферментов и увеличение окислительных и гидролитических. Кроме снижения уровня протеолипидов, составляющих структуру миелиновой оболочки проводящих путей, демиелинизации, патологии собственно липидов в пораженных путях не выявлено. Патология со стороны внутренних органов: кардиомегалия с гипертрофией миоцитов, а в поджелудочной железе хронический интерстициальный фиброз и воспалительная инфильтрация.
Клиническая картина.
Первые симптомы заболевания возникают обычно на 1—2-м десятилетии жизни, чаще всего в препубертатном периоде, характеризуются незаметным появлением симптомов, относительно быстрым прогрессированием процесса и сочетанием типичных неврологических и экстраневральных клинических проявлений.
Неврологические симптомы.
Заболевание обычно манифестирует появлением неловкости, неуверенности при ходьбе, особенно в темноте (признак заднестолбовой атаксии), больные начинают пошатываться, часто спотыкаются. Вскоре к атаксии при ходьбе присоединяются дискоординация в руках, изменение почерка, слабость в ногах. Уже в самом начале заболевания может отмечаться дизартрия. Речь приобретает характер стаккато или эксплозивности (взрывчатости) в результате несогласованности дыхания и фонации. Ранним и важным дифференциально-диагностическим признаком болезни Фридрейха является исчезновение сухожильных и надкостничных рефлексов. Угнетение рефлексов (в первую очередь ахилловых и коленных) может на несколько лет опережать манифестацию других симптомов болезни и быть самым ранним проявлением неврологической дисфункции. В развернутой стадии заболевания у больных обычно наблюдается тотальная арефлексия. Как писал в 1975 г. J.Tyrer, «…многие неврологи будут как минимум колебаться в постановке клинического диагноза атаксии Фридрейха при наличии оживленных сухожильных рефлексов». Специфичность сухожильной арефлексии связана с тем, что первичной мишенью дегенеративного процесса при атаксии Фридрейха являются чувствительные нейроны спинномозговых ганглиев, переключающие сигналы от мышечных веретен и составляющие важное звено спинального рефлекса на растяжение.
Типичным неврологическим проявлением болезни Фридрейха является нарушение глубокой (суставно-мышечной и вибрационной) чувствительности. Довольно рано у больных при неврологическом осмотре может быть обнаружен симптом Бабинского, мышечная гипотония. По мере прогрессирования заболевания постепенно нарастают мозжечковая и сенситивная атаксия, слабость и атрофия мышц ног. Атаксия вызвана комбинацией церебеллярной асинергии и поражением заднего столба с его чувствительными проводниками. Она обычно более выражена в ногах, чем в руках, и выявляется при исследовании походки и статики ребенка.
В поздней стадии болезни парезы, амиотрофии и расстройства глубокой чувствительности распространяются на руки. Больные перестают самостоятельно ходить и обслуживать себя из-за глубокого распада моторных функций. В ряде случаев наблюдается нистагм, снижение слуха, атрофия зрительных нервов; при длительном течении болезни отмечается нарушение функции тазовых органов. По поводу деменции мнения противоречивы. Если у взрослых она описана, то у детей умственная отсталость и деменция встречаются редко.
Экстраневральные симптомы.
Среди экстраневральных проявлений болезни Фридрейха необходимо выделить поражение сердца, которое встречается более чем у 90 % больных. Характерным является развитие типичной прогрессирующей кардиомиопатии. Кардиомиопатия преимущественно носит характер гипертрофической, но в отдельных случаях возможно развитие дилатационной кардиомиопатии. Не исключено, что эти изменения сердца при болезни Фридрейха являются различными стадиями одного процесса. Кардиомиопатия проявляется болями в области сердца, сердцебиением, одышкой при физической нагрузке, систолическим шумом и другими симптомами. Более чем у половины больных кардиомиопатия является непосредственной причиной смерти. Другим характерным экстраневральным проявлением болезни Фридрейха являются скелетные деформации: сколиоз, «стопа Фридрейха» (высокий вогнутый свод стопы с переразгибанием пальцев в основных фалангах и сгибанием в дистальных), деформация пальцев рук и ног и др. Эти нарушения также могут появляться задолго до развития первых неврологических симптомов. Следует отметить, что, несмотря на широкое распространение эпонимического термина «стопа Фридрейха», данный тип деформации стоп не является патогномоничным для болезни Фридрейха и встречается при некоторых других дегенеративных заболеваниях нервной системы, например при невральной амиотрофии Шарко — Мари, спастической параплегии Штрюмпеля и др. К экстраневральным проявлениям болезни Фридрейха относятся эндокринные расстройства (сахарный диабет, гипогонадизм, инфантилизм, дисфункция яичников).
Патология со стороны органа зрения при атаксии Фридрейха встречается относительно редко и может включать аномалии рефракции, пигментную ретинопатию, катаракту (De Silva R. et al., 1998).
Принято считать, что экстраневральные признаки болезни Фридрейха — это проявление плейотропного действия одного мутантного гена.
Диагностика
— При исследовании вызванных зрительных потенциалов выявляются генерализованное снижение амплитуды потенциалов и удлинение времени их появления. Уменьшение амплитуды, вероятно, является следствием утраты функционирующих волокон в зрительном тракте. Результаты электроретинограммы обычно нормальные.
— Соматосенсорные вызванные потенциалы, регистрируемые от надключичных отведений, отличаются от нормальных уже на самых ранних стадиях болезни, но они не сопровождаются снижением проводимости по периферическому нерву.
— При молекулярно-генетическом обследовании пациентов с клинически типичными проявлениями БФ на увеличение тринуклеотида ГАА расширение аллеля обнаруживается не у всех. При этом возможна точечная мутация или делеция в гене БФ на обеих хромосомах. В этих случаях это может быть фенокопия БФ, так как увеличение триплета было описано у многих больных атипичной атаксией и у пациентов с генерализованной хореей.
— МР-томография позволяет уже в ранней стадии болезни визуализировать атрофию спинного мозга, уменьшение поперечного размера спинного мозга, особенно усиливающееся в каудальном направлении, а при более длительном течении — умеренно выраженную атрофию продолговатого мозга, моста и мозжечка.
— Компьютерная томография головного мозга имеет ограниченное значение (в связи со спинальной локализацией основных морфологических изменений). В большинстве случаев при КТ обычно обнаруживается либо слабая степень атрофии мозжечка, либо отсутствие изменений. Лишь в поздней стадии заболевания при КТ можно выявить ряд изменений: атрофию полушарий и червя мозжечка, расширение IV желудочка, стволовых цистерн, боковых желудочков и субарахноидального пространства больших полушарий.
— Электрофизиологические исследования — электронейромиографический паттерн (отсутствие или значительное снижении амплитуды потенциалов действия чувствительных нервов конечностей при сравнительно небольшом снижении скорости проведения импульса по двигательным нервам.)
— Электрокардиография и эхокардиография – выявление кардиомиопатии, (нарушение ритма, инверсия зубца Т, изменения проводимости). Особенно часто отмечаются нарушения проводимости, вплоть до полной блокады, и гипертрофия межжелудочковой перегородки. В ряде случаев клинические и электрокардиографические симптомы поражения сердца иногда на несколько лет опережают появление неврологических нарушений.
— Исследование содержания глюкозы в крови с проведением специальных нагрузочных тестов толерантности к глюкозе (для исключения сахарного диабета).
— Рентгенологическое исследование позвоночника (характеристика костных деформаций).
— Для оценки митохондриальных нарушений с помощью цитохимического метода наиболее целесообразны представляется определение активности ряда ферментов-дегидрогеназ лимфоцитов: сукцинатдегидрогеназы (СДГ),
а-глицерофосфа дегидрогеназы (ГФДГ) глутаматдегидрогеназы (ГДГ), лактатдегидрогеназы (ЛДГ), малатдегидрогеназы (МДГ) и др. Выявляется их достоверное снижение.
Критерии диагноза
Основными критериями диагноза болезни Фридрейха являются:
1. аутосомно-рецессивный тип наследования;
2. дебют в подростковом, реже в юношеском возрасте;
3. атаксия, арефлексия, нарушение глубокой чувствительности, слабость и атрофии мышц ног, позднее рук;
4. экстраневральные симптомы:
а) скелетные деформации: сколиоз, полая стопа («стопа Фридрейха»), деформация пальцев ног и рук и др.;
б) эндокринные расстройства: сахарный диабет, гипогонадизм, инфантилизм, дисфункция яичников;
в) кардиомиопатия (гипертрофическая, реже дилатационная): изменения на ЭКГ и ЭхоКГ;
г) катаракта;
5. атрофия спинного мозга, визуализирующаяся на МР-томо- граммах;
6. ДНК-диагностика. Диагноз подтверждается определением размера повторов ГАА.
Дифференциальный диагноз.
Болезнь Фридрейха необходимо дифференцировать, в первую очередь, со второй по частоте прогрессирующей атаксией с началом в детском возрасте — атаксией-телеангиэктазией (болезнью Луи-Бар). Клинически она отличается наличием на коже телеангиэктазий (чрезмерное локальное расширение мелких сосудов, преимущественно прекапилляров и капилляров), отсутствием скелетных аномалий, частыми и тяжело протекающими инфекциями дыхательных путей, отсутствием или крайне низким уровнем IgA, высоким уровнем а-фетопротеина. На МРТ выявляется гипоплазия мозжечка, чаще его червя.
От клинически весьма сходной формы наследственной атаксии, вызываемой дефицитом витамина Е, и близким к ней синдромом Бассена—Корнцвейга. Для дифференциальной диагностики необходимо определять содержание в крови витамина Е, исследовать липидный профиль крови, мазок крови на наличие акантоцитоза.
Следует также исключать обменные заболевания, наследуемые по аутосомно-рецессивному типу и характеризующиеся нередко развитием спиноцеребеллярной атаксии — Gm 1- и Gm 2-ганглиозидоз и галактосиалидоз (исследование активности І-галактозидазы и гексозаминидазы А), болезнь Краббе (исследование фермента галактозилцерамидазы), поздний вариант болезни Ниманна—Пика (определение содержания сфингомиелинов цереброспинальной жидкости, исследование стернального пунктата на наличие «пенистых» клеток).
Дифференциальный диагноз болезни Фридрейха и рассеянного склероза обычно не вызывает затруднений, поскольку для последнего нехарактерны такие симптомы, как сухожильная арефлексия, мышечная гипотония, амиотрофии, экстраневральные проявления, а также в связи с отсутствием при болезни Фридрейха ремиссий и очаговых изменений плотности вещества мозга при КТ и МР-томографии.
Дифференциальный диагноз проводят также с сифилитическим поражением ц.н.с., опухолями мозжечка, фуникулярным миелозом.
Лечение.
В связи с недостаточностью энергопродукции при болезни Фридрейха важное значение в лечении данного заболевания в настоящее время придается препаратам, поддерживающим функцию митохондрий и широко применяющимся в терапии других митохондриальных болезней.
Общий принцип лечения данными препаратами состоит в сочетанном назначении лекарств, синергично влияющих на разные уровни энергетического метаболизма. Рекомендуется одновременное назначение как минимум трех лекарственных средств из первых трех групп. (Препараты, повышающие активность дыхательной цепи митохондрий, кофакторы энзимных реакций энергетического обмена, антиоксиданты). Полученные предварительные результаты свидетельствуют о том, что вышеуказанный комплекс митохондриальных препаратов может способствовать определенной стабилизации энергетических процессов в клетке и тем самым в какой-то степени замедлять развитие нейродегенерации при атаксии Фридрейха.
Комплексное лечение пациентов должно включать применение довольно широкого спектра симптоматических средств. Обычно назначают препараты, улучшающие метаболизм миокарда: рибоксин, кокарбоксилазу, предуктал и др.
Пациенты чувствуют себя лучше при ограничении углеводов в пище до 10г/кг, поскольку высокое их потребление является своеобразной «провокацией», усиливающей дефект энергетического обмена.
Дети с БФ могут оставаться активными максимально долго, занимаясь лечебной физкультурой и комплексами корректирующих упражнений, которые следует фокусировать на тренировку баланса и силы мышц. При такой программе упражнений не развивается кардиомиопатия.
Ортопедическое хирургическое лечение скелетных деформаций, особенно прогрессирующего сколиоза, который является главной причиной болей, может быть необходимым, если ношение ортопедического корсета не предотвращает развитие сколиоза. Проводится общеукрепляющее лечение (витамины), а также препараты, влияющие на тканевый обмен (пирацетам, аминалон, ацефен, церебролизин), лечение которыми следует периодически повторять.
Прогноз.
Болезнь Фридрейха характеризуется неуклонно прогрессирующим течением, длительность болезни может варьировать в широких пределах, но чаще всего не превышает 20 лет.
Непосредственными причинами смерти могут быть сердечная и легочная недостаточность, инфекционные осложнения.
Профилактика БФ основана на медико-генетической консультации.
Болезнь Фридрейха до наших дней остается, пожалуй, наиболее четко очерченным синдромом в обширном и гетерогенном ряду атаксий; это, бесспорно, свидетельствует об исключительном таланте исследователя, именем которого названо данное заболевание.
Николаус Фридрейх умер от разорвавшейся аневризмы аорты в 1882 году в возрасте 57 лет.
В своем трактате, посвященном его преподавателю и наставнику Рудольфу Вирхову, он писал: «Для меня, как для клинициста принципы клеточной патологии стали полярной звездой в лабиринте патологического процесса».
Список использованной литературы
1. С.Н. Иллариошкин. Наследственные атаксии и параплегии. — «Медицина», 2006.
2. К.Г. Арзуманова, диссертация «Спинно-церебелярные дегенерации», 1990.
3. Ю.Е. Вельтищев, П.А.Темин. Наследственные болезни нервной системы. «Медицина», 1998.
4. М.Б. Кроль. Е.А. Федорова. Основные невропатологические синдромы. «Медицина», 1966.
5. А.С. Петрухин. Неврология детского возраста. – «Медицина», 2004.
6. П.А. Темин, Л.З.Казанцева. Наследственные нарушения нервно-психического развития детей. Руководство для врачей. «Медицина», 2001.
7. «Журнал неврологии и психиатрии им.С.С.Корсакова» №4 1996.
8. Ф. Блум, А. Лейзерсон, Л. Хофстедтер. Мозг, разум и поведение, «Мир», 1998.
9. Д.Р.Штульман, О.С. Левин «Неврология. Справочник практического врача». Издательство «МЕДпресс-информ», 2005г.
10.Журнал Science, 8.03.1996, “Friedreich’s Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet Repeat Expansion.